It takes 10-15 years and over $2 billion on average to bring a single drug to approval. This is driven by evidentiary requirements — several phases of clinical trials, each requiring greater enrollment of patients — and the failures along the way.
In February 2026, Center for Biologics Evaluation and Research (CBER) Director Vinay Prasad and Food and Drug Administration (FDA) Commissioner Martin Makary published a commentary/opinion piece in the New England Journal of Medicine titled “One Pivotal Trial, the New Default Option for FDA Approval — Ending the Two-Trial Dogma.” The article closes noting that the Food and Drug administration’s (FDA) new default position is “one adequate and well-controlled study, combined with confirmatory evidence, will serve as the basis of marketing authorization of novel products.”
This latest information reflects long-standing debates about trial design efficiency, evidentiary and data sufficiency, and the balance between rapid access and robust evidence for novel medications. Prasad and Makary suggest that this will lead to cost and time savings, resulting in a “surge in drug development.”
Industry response has been generally positive, with reactions ranging from transformative to cautious optimism. However, former Center for Drug Evaluation and Research (CDER) Director Richard Pazdur has called this “very dangerous,” highlighting that “these [end points] may be subject to bias, and sometimes it’s important to have clinical trials and duplications of clinical trials in those areas. My caution is, all that glitters is not gold, one has to be very careful, and I hope the upcoming guidance that is being written on this is cognizant of the differences that would exist between other therapeutic areas.”
With the departures of Makary and Prasad, it is unclear if this will become the new standard. If it does, this change could be seen as a win for biopharma, although uncertainties remain.
In this edition of Executive Insights, L.E.K. Consulting contextualizes the news and provides key questions that biopharma executives should be asking.
How big of a shift is this? What drugs are impacted?
Under Title 21 of the Code of Federal Regulations, FDA approval requires “substantial evidence” of effectiveness. Historically, this has often been interpreted as evidence derived from two adequate and well-controlled studies. However, the statute does not explicitly mandate two trials.
Precedent has allowed approval based on a single adequate and well-controlled study with confirmatory evidence in appropriate circumstances. This distinction is important, and the commentary from Prasad and Makary does not alter the statutory standard but reframes how substantial evidence may be demonstrated in modern development settings.
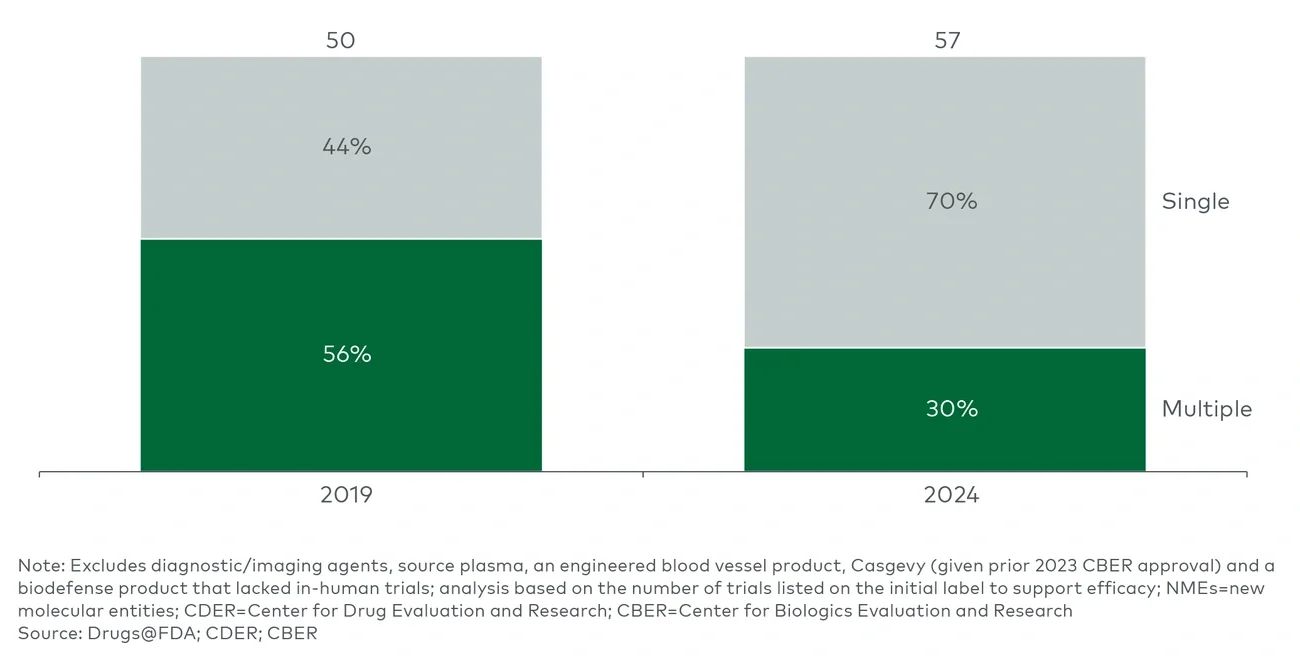
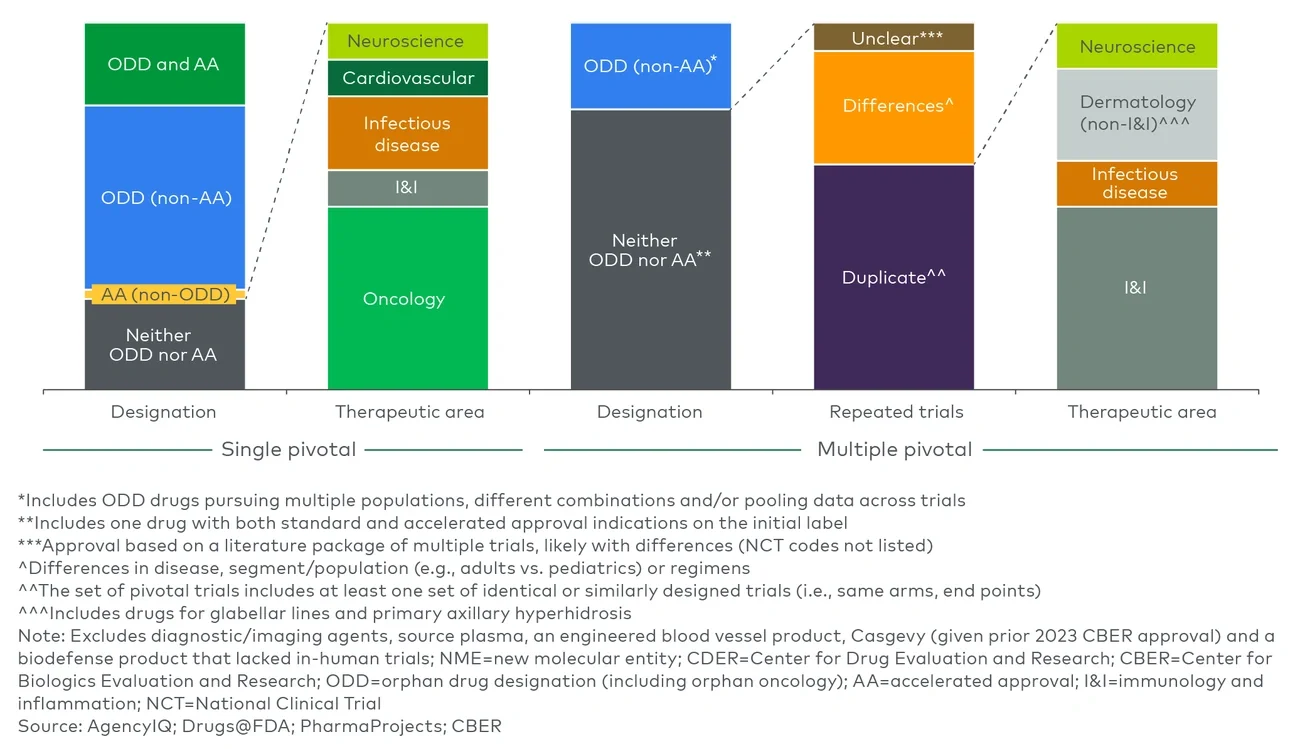
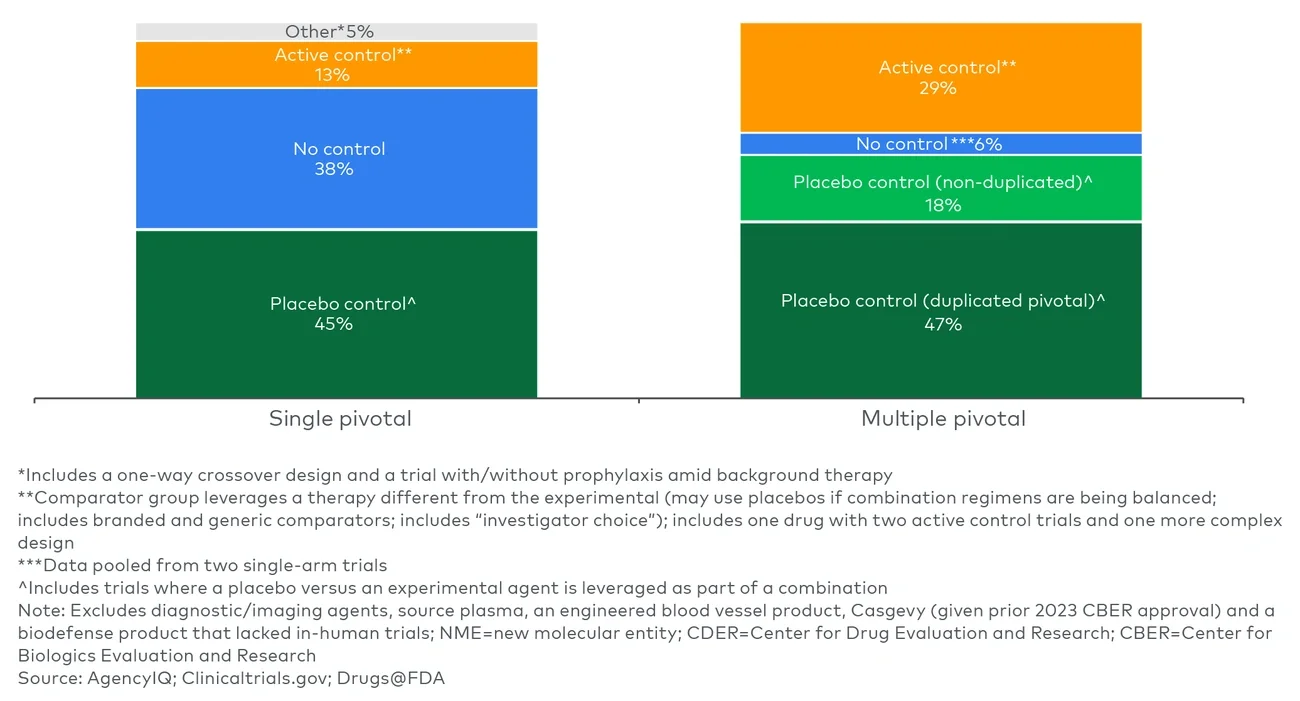
Looking at approvals over time before the Trump administration, a declining number of therapeutic approvals have required multiple efficacy trials to support approval, with 60%-70% of CDER and CBER new molecular entity (NME) approvals in 2024 relying on a single trial (see Figure 1).