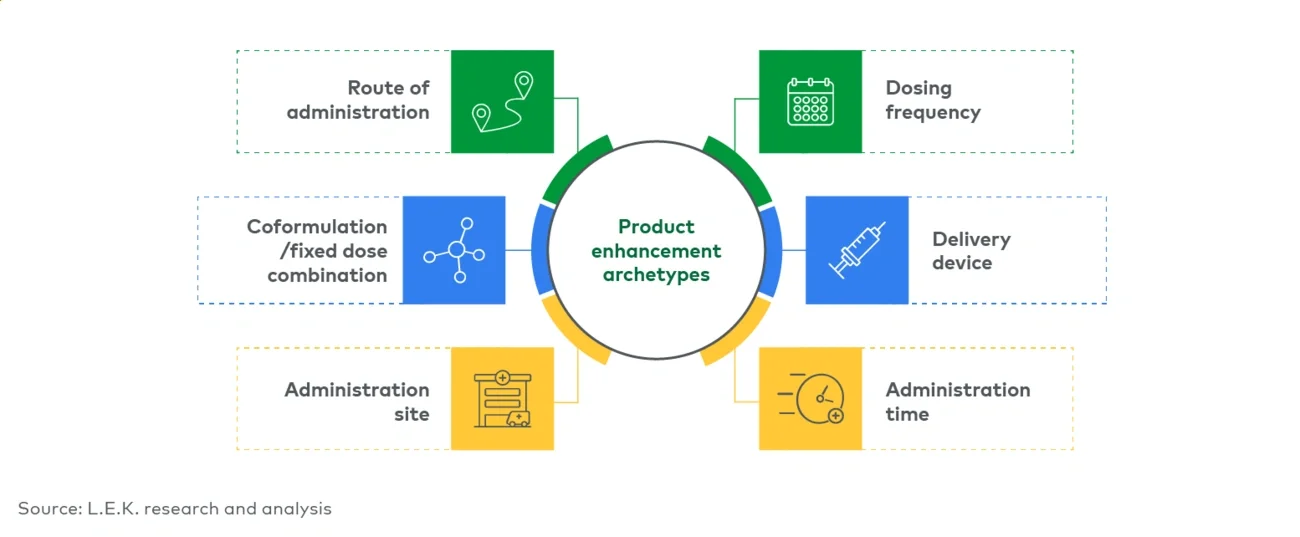
Altering the dosing frequency can have implications across stakeholders, making it critical to ensure the value proposition is optimized. Key questions include:
- Is dosing frequency a burden impacting patient compliance or persistence?
- How does the dosing schedule align with physician check-ins?
- Is dosing standardized in practice or do physicians opt to titrate?
- Is there a chance a patient will need to switch medications during the dosing window?
- How can a seamless transition be enabled?
- How do the dosing frequency and pricing impact physician economics and operations (e.g., inventory management, patient throughput)?
Delivery device
Ideally, delivery devices ease administration, minimizing human error and/or improving process flows. These improvements can help enable at-home administration for drugs that might otherwise require an in-office visit while allowing administration that accommodates the patient’s schedule. To illustrate the concept simply, a prefilled syringe removes a process step and potential error associated with vial transfer. An autoinjector may further improve the process, allowing a patient to self-inject with a hidden needle and improved ergonomics. The Auvi-Q autoinjector provides voice-guided instructions, aiding emergency epinephrine administration. A manufacturer can also offer dosing optionality through a variety of delivery devices. Improving on drug delivery may provide additional benefits as well. Having multiple device options provides optionality; for example, Empaveli is a large-volume SubQ drug that can be administered with a commercially available pump (e.g., Koru) or an on-body delivery system (e.g., Enable’s enFuse).
Sometimes a device can change a process flow more radically — Neulasta OnPro provides a delayed dose, obviating the need for a next-day clinic visit and addressing a market pain point in a way that poses a strong defense against biosimilar competition. Drug delivery may also incorporate digital features and mobile connectivity. For example, the Omnipod automated insulin delivery system captures disease management insights and integrates with continuous glucose monitoring devices (e.g., Dexcom, FreeStyle Libre). For additional perspectives on drug delivery devices, please see other recent L.E.K. Executive Insights.
When opting for a delivery device, biopharmas need to understand the range of administrators (patient, caregiver, healthcare provider [HCP]), the strength of the value proposition for each and any potential risks. Human factor studies are critical. Key questions include:
- Is the device essential or a nice-to-have?
- How would the device improve drug delivery?
- How valuable is this improvement to different user archetypes?
- What are the risks of misuse or mechanical failure?
Administration time
Delivery devices and IV-to-SubQ conversions — both discussed above — are among the most impactful levers for reducing administration time and improving patient convenience. For HCP-administered drugs, this can help improve throughput, allowing centers to dose more patients given the constraints on infusion chairs and/or support staff. Such benefits need to be kept in the context of real-world drug usage and administration to ensure the value proposition is not diluted. Key questions include:
- Is the drug dosed in combination with other IVs?
- What does the HCP need to do to administer the drug?
- Will new process flows be required?
Administration site
Shifting the site of administration may drive benefits but requires navigating a more complex web of reimbursement economics and stakeholder incentives.
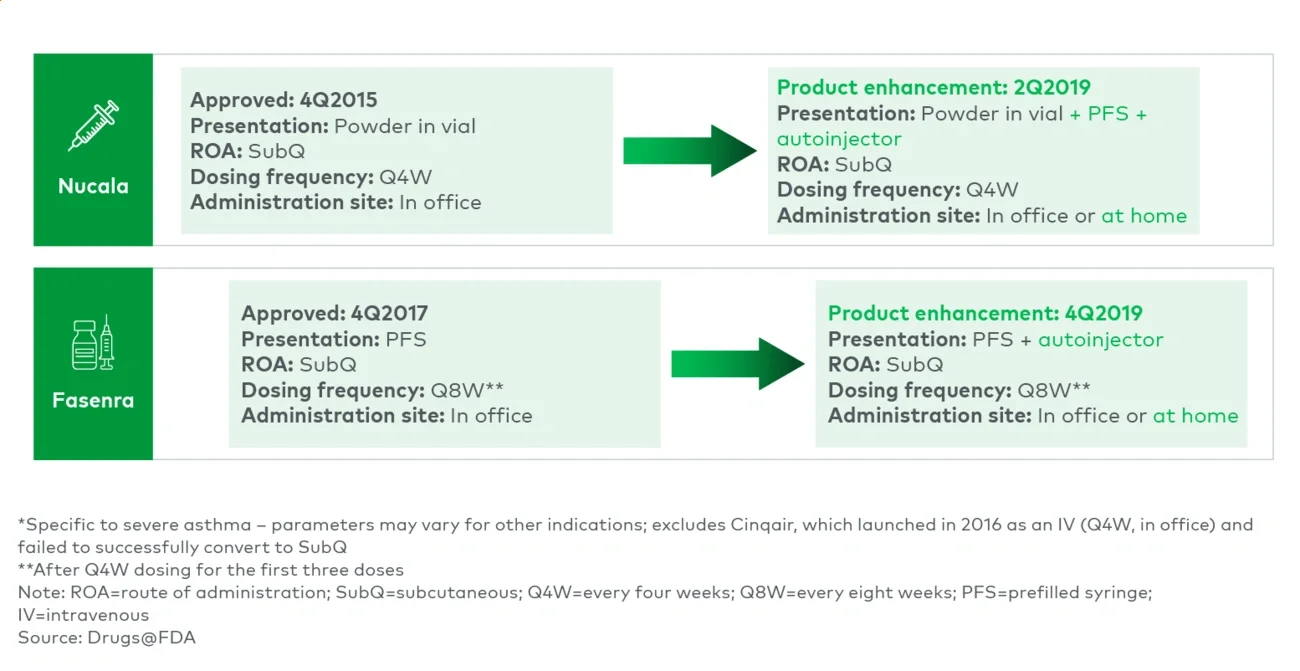
Moving from outpatient HCP administration to self-/caregiver administration improves convenience — patients no longer must travel to receive their medication. It also shifts a product from the medical benefit to the pharmacy benefit. This provides payers with greater utilization management control and financial incentives (as such, sometimes payers push toward “white bagging” to circumvent the medical benefit). For HCPs, the impact is more mixed. It frees up HCP time and resources (e.g., inventory, administration); however, it removes the buy-and-bill financial incentives, it may remove a desired patient check-in and it may introduce dosing errors.
Examples include Nucala and Fasenra (initially HCP-administered SubQs) and Entyvio (IV to SubQ). Neulasta OnPro’s delayed dose allowed it to remain HCP-administered and reimbursed under medical benefit, retaining buy-and-bill economics while still enabling at-home dosing. Conversely, Cosentyx launched in 2015 as a self-administered SubQ and added an IV option in 2023, which allowed it to address patients who preferred not to self-administer and physicians/centers that preferred buy-and-bill.
Biopharmas can also help shift clinical practice to move HCP-administered inpatient (i.e., hospital benefit) drugs to outpatient. Chimeric antigen receptor T (CAR-T) therapies are a prime example, with studies supporting outpatient dosing, with inpatient admission as needed. This frees up hospital resources while making the billing/reimbursement simpler and more financially viable.
Key questions when altering administration site include: How does in-office administration impact patient compliance and fit into disease management? How do prescribers value the buy-and-bill option? What is the expected impact on utilization management?
Coformulation/fixed-dose combination
Drugs may be coformulated or combined with other agents commonly dosed together individually (e.g., Janumet is Januvia plus metformin) or to enable a new dosing option (e.g., hyaluronidase-based formulations enabling IV-to-SubQ conversion). Phesgo is an example that does both — a combination of Roche/Genentech’s Herceptin, Perjeta and hyaluronidase for SubQ use. A manufacturer can also opt to introduce a new product in fixed-dose combination with an older one it owns (e.g., Opdualag), providing life cycle benefits for the older product. A fixed-dose combination acts as a new branded product and comes with exclusivity benefits. For a manufacturer, the cost of a fixed-dose combination varies and may become operationally complex if spanning multiple branded owners. Key questions when developing a fixed-dose combination include:
- Are the components’ dosing and schedules conducive to coformulation?
- Are there any other combination agents that need to be added on top?
- What is the risk of circumventing the fixed-dose combination with individual components?
Others
The categories above are not exhaustive. A few additional examples illustrate how product improvements often span multiple factors simultaneously. Humira removed citrate, lowered dosing volume and altered needle gauge to reduce injection-site pain. Daybue was originally launched as an oral solution that required refrigeration and discarding 14 days after opening the bottle; Daybue Stix, an oral powder, was introduced to offer flexibility and choice in dose volume and taste (by mixing with different water-based liquids) while allowing for room-temperature storage.
Biopharmas should carefully plan for optimized product improvements across the drug life cycle
Investment in life cycle management behind a strong product is a critical lever, allowing products to reach new patients, enhance benefits to users and protect share from branded and generic competition. Given the breadth of formulation and delivery options available, biopharmas should consider the following guiding principles:
- Do not sacrifice time to market: Biopharmas should maintain their focus on bringing viable products to market in a timely fashion, accelerating patient benefit and generating revenues to support operations. Unless the initial product presentation is a significant detraction, product improvements should be made after the initial approval and launch.
- Plan product improvements in parallel with development: Given pressures on product life cycle (e.g., Medicare Price Negotiation, competition), biopharmas should plan ahead. Sequential development may sacrifice time on market and the ability to convert patients from one product to another.
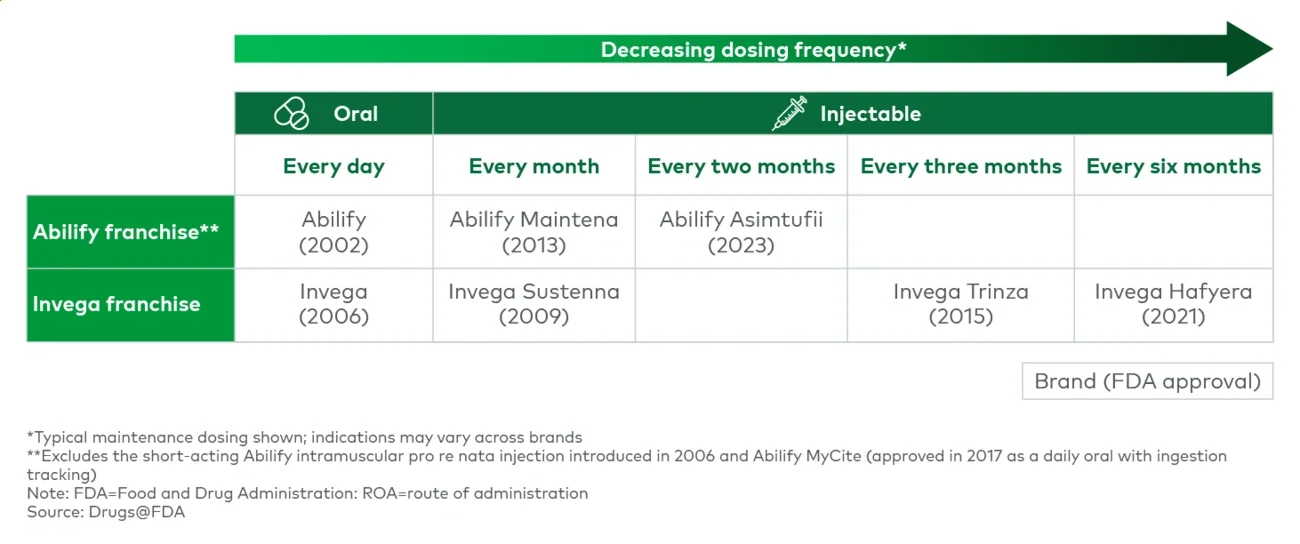
- Enable switching: Generate data (e.g., switch studies, noninferiority/head-to-head, human factors), create protocols (e.g., Invega’s) and offer support services to motivate switching to the next-gen drug while alleviating any concerns or frictions from physicians, patients or payers. An ideal strategy migrates ahead of loss of exclusivity.
- Optimize product improvements to address stakeholder preferences and unmet needs: It is critical to understand the implications of planned product improvements across stakeholder types, optimizing the improvement and its commercial support. Often the theoretical benefit is more nuanced in the real world. Value proposition testing should be coupled with a detailed understanding of practice economics, which should be incorporated in the go-to-market strategy.
- Ensure regulatory and legal integrity and distinguish genuine value from evasion: The policy environment around life cycle management is tightening, not stabilizing. CMS is moving to codify the Medicare Drug Price Negotiation Program permanently and to narrow the fixed-combination and new-formulation pathways that have been used to defer selection, and the Federal Trade Commission has signaled continued scrutiny of product/ patent hopping and patent thickets. Enhancements that deliver real, demonstrable benefit to patients, physicians or payers (e.g., improved adherence, reduced administration burden, lower total cost of care) sit on durable ground and are unlikely to draw the same scrutiny. Enhancements pursued primarily to reset exclusivity or evade negotiation carry escalating regulatory, legal and reputational risk, and biopharmas should pressure-test each planned improvement against that distinction, not merely confirm technical compliance.
L.E.K. has extensive experience optimizing product value propositions and supporting life cycle development planning across the biopharma sector. For more information, please contact us.
L.E.K. Consulting is a registered trademark of L.E.K. Consulting LLC. All other products and brands mentioned in this document are properties of their respective owners. © 2026 L.E.K. Consulting LLC