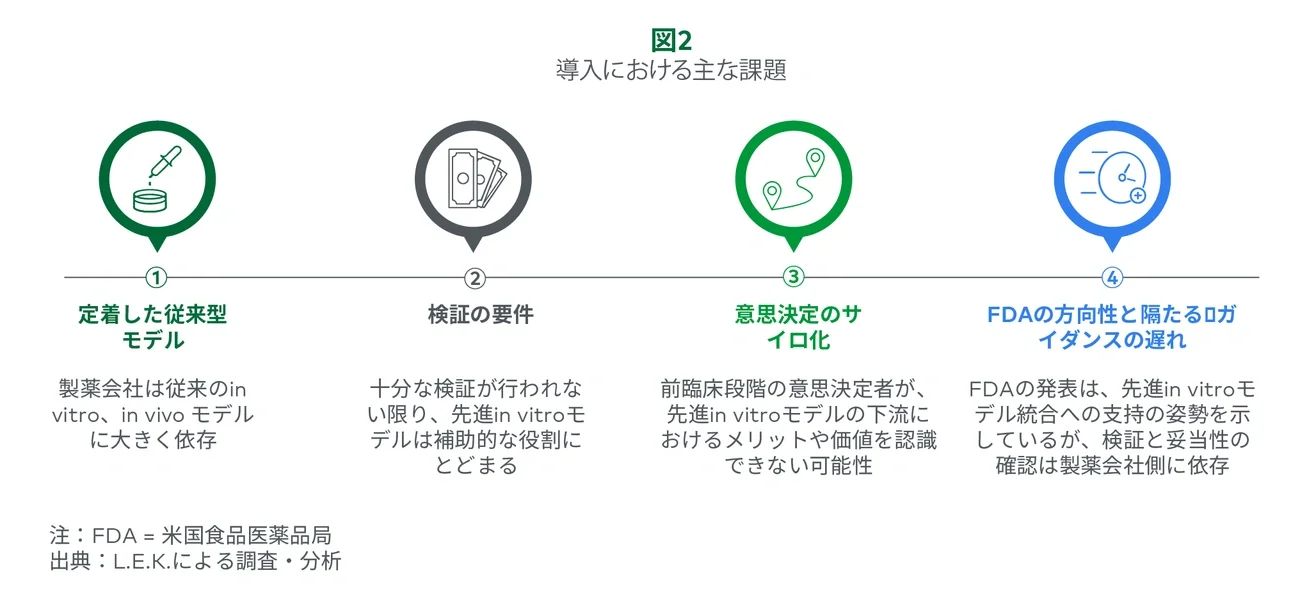
1. Current in vitro and in vivo models are highly entrenched within discovery and preclinical development workflows.
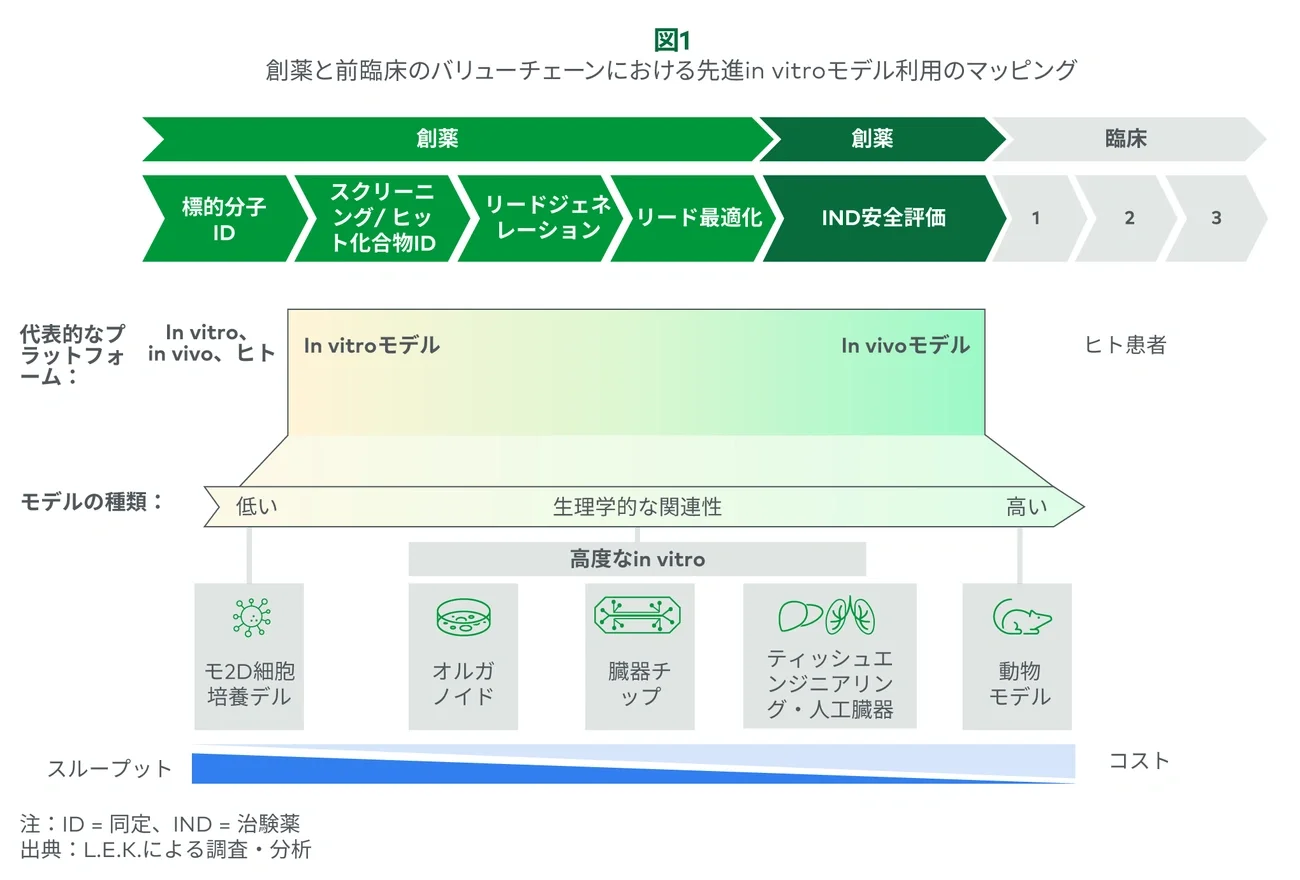
In vitro and in vivo models have long been foundational to pharmaceutical development, not because they are perfect but because they are trusted, are practical, and have consistently supported the discovery of clinically successful drugs. In vitro models typically consist of 2D cell models, and while these models are simplistic, pharma leverages them in high-throughput formats (384-well plates) in single-readout assays to assess efficacy or safety across large compound libraries.
Often these 2D cell model assays recapitulate a disease state (e.g., breast cancer) or test a safety attribute (e.g., hERG ion channel safety). Although advanced in vitro models may offer greater accuracy, the acceptance, entrenchment and throughput of 2D models create a high barrier to adoption.
Additionally, the higher cost of these advanced in vitro systems may not be justified by their performance benefits, making practicality a key consideration. Recently, suppliers have focused on complex disease areas that lack a simple 2D model (e.g., I/O or muscular dystrophy) and where advanced models support a higher throughput not previously possible. Suppliers are increasingly integrating AI with advanced models to drive throughput and extract insights about disease mechanisms. AI and machine learning (ML) algorithms can help simplify complex data and identify early patterns of efficacy and toxicity.
In vivo rodent models are typically used starting in lead optimization to evaluate candidates in physiologically relevant systems. These models are leveraged for a suite of experiments (e.g., safety, efficacy and ADME studies), often with multiple readouts from a single animal. For example, dose escalation studies will also enable toxicity and pathological assessment of organs (e.g., liver, kidney, heart).
While advanced in vitro models may be as predictive as in vivo models, their lack of physiological complexity/context (e.g., multi-organ systems) limits widespread utilization and has kept many researchers from replacing in vivo models. Animal models that are resource-intensive or offer limited translational value (e.g., nonhuman primates) are increasingly viewed as entry points for advanced in vitro approaches, and recently, suppliers have been targeting use cases where large numbers of animals are utilized for only a single specific readout (e.g., gene therapy, titer optimization).
Suppliers are using AI/ML in combination with these systems to further help researchers model vector performance, predict optimal capsid design and understand transduction efficiency. Beyond gene therapy, AI and advanced systems are helping reduce animal use across complex and poorly predicted areas, such as cardiac toxicity and immune response, demonstrating that advanced systems may serve as a strong starting point toward supporting the FDA’s goal of reducing and eventually eliminating animal testing.
2. Advanced in vitro systems are likely a supplemental cost until fully validated.
Validation is key to convincing stakeholders to choose an advanced in vitro model over the current gold standard; however, it remains a moving target. Without sufficient validation, researchers must still conduct legacy experiments, making advanced in vitro models a supplemental cost rather than a replacement.
Stakeholders often require both retrospective and prospective validation. For retrospective validation, suppliers leverage therapeutics with known toxicity profiles to show that advanced in vitro models can predict similar specificity and sensitivity. Some models have shown toxicity from therapeutics that failed clinical trials but were not deemed toxic using traditional in vitro and in vivo models.
Pharmaceutical stakeholders also seek prospective validation, where other researchers successfully use this advanced in vitro tool to support a drug’s progression to clinical trials and potentially eventual approval. Taken together, retrospective and prospective validation are costly, as they require high upfront investments and initial adoption by multiple champions.
In today’s cost-constrained and time-sensitive environment, studies are often hard to justify, particularly when they do not replace existing experiments. Justifying these extra costs remains a headwind for suppliers, and without broad validation, advanced in vitro models are likely to remain additive to development workflows. To overcome this, suppliers must sharpen their value proposition to attract champions and clearly demonstrate practical utility.
A growing number of suppliers are leveraging AI/ML to validate and benchmark their models against known clinical outcomes to enhance regulatory credibility and commercial adoption. AI/ML algorithms can analyze high-dimensional data (e.g., transcriptomics, phenotypic screens) to demonstrate that in vitro models align with relevant disease biology and drug response. The FDA’s most recent guidance may accelerate broader collaboration across advanced in vitro approaches to build confidence and momentum.
3. Advanced in vitro model value accrues downstream and may go unrecognized by purchasing stakeholders
Discovery and preclinical teams are often siloed from their clinical counterparts, hindering the realization of advanced in vitro models’ full value. This disconnect means the buying team may not recognize downstream benefits, such as improved PTRS, from better early-stage decisions. For example, if an organoid model helps discovery scientists eliminate candidates with hepatotoxic profiles, the benefit of delivering a lead candidate with a lower risk of liver damage to preclinical studies or clinical trials may go unnoticed.
This lack of cross-team visibility also applies to cost and time savings, as preclinical and clinical teams may not fully recognize the impact of early discovery failures. These efficiency gains and cost savings can be overlooked when transitioning candidates across stages of the value chain. Additionally, “kill quickly” is not always incentivized, as many researchers are evaluated based on the number of candidates, not necessarily the success of those candidates downstream.
To demonstrate value, advanced in vitro model suppliers must track how their tools have supported key decision points (e.g., deprioritizing candidates based on early safety or efficacy signals) and highlight the productivity gains (e.g., longer and more costly in vivo studies). This task can be difficult, as each clinical asset likely passes through multiple models and hundreds of experiments, and each advanced in vitro model must lean on its value proposition to convince the pharma industry of its impact.
4. The FDA’s recent announcement signals progress, but broad replacement of animal models will be a cautious and slow process.
Despite the growing evidence supporting advanced in vitro models over traditional in vitro and in vivo testing, the FDA has been slow to fully embrace these technologies and integrate them into regulatory frameworks. While the agency has taken incremental steps in recent years, such as issuing guidance documents, supporting legislation (e.g., FDA Modernization Act 2.0), launching qualification programs (e.g., ISTAND) and supporting collaborative research, the pace of adoption remains cautious.
The most recent FDA guidance indicates a more active stance on reducing animal usage and relying on alternative models. However, beyond the reduction of primate study requirements for mAbs, the roadmap remains aspirational.
Outside select, more mature applications (e.g., liver OOC), the advanced in vitro space is nascent, with many emerging models lacking validation and the confidence of sponsors. The FDA highlights early-stage concepts, such as whole-body-on-a-chip systems and AI/ML models for pharmacokinetics, but provides limited clarity on regulatory expectations or actionable pathways for adoption.
While the FDA’s tone is supportive of a transition to more predictive tools, for now the burden of validation and regulatory evidence falls to pharma sponsors, which must determine whether the FDA is truly accepting of data from organoids or OOC systems. Additionally, the FDA and the pharma industry must account for other stakeholders such as clinical teams that may be hesitant to recruit for studies built on unfamiliar toxicology data.
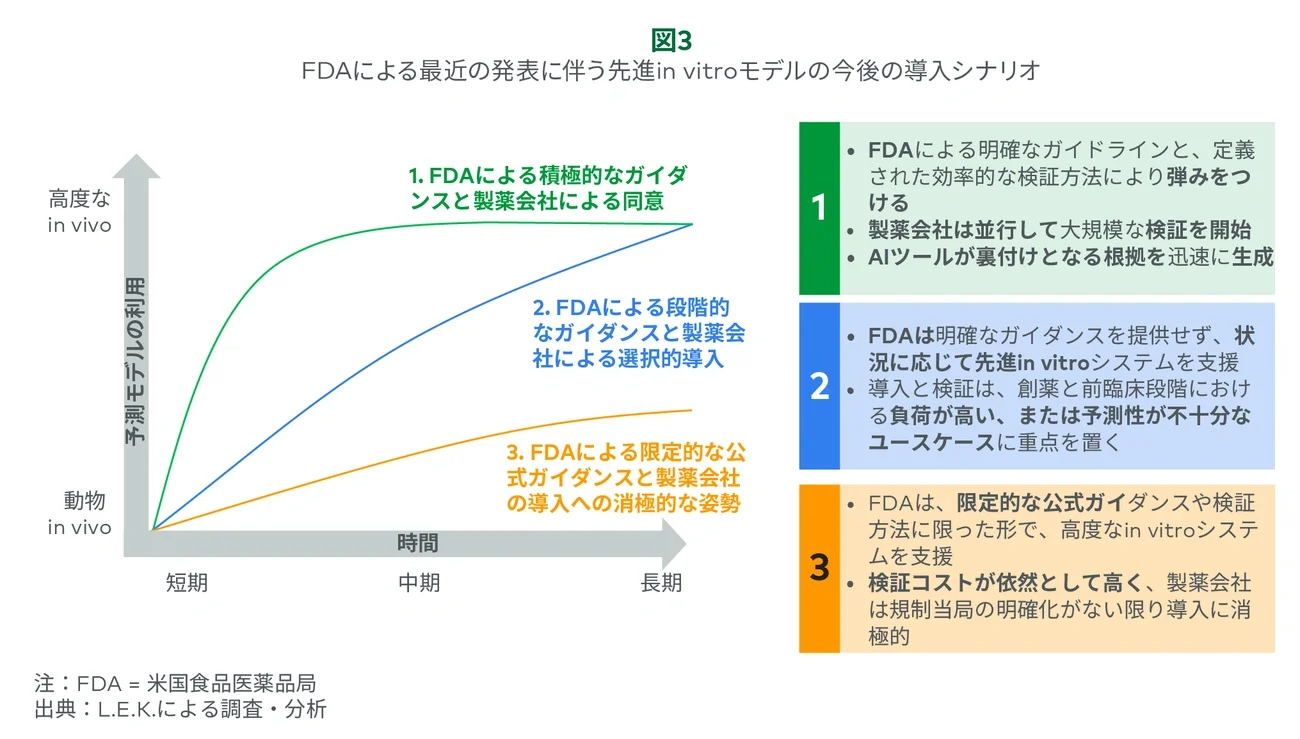
Still, this guidance marks an important inflection point. Depending on how industry and regulators act in the near, mid- and long term, adoption could accelerate meaningfully or continue at a slower pace (see Figure 3). Pharma will play a critical role in generating the validation data needed to shift regulatory perception, especially in high-priority use cases where existing models are costly or poorly predictive.
If adoption is effectively executed with coordinated validation, clearer expectations and support across advanced in vitro, in silico and AI/ML models, this recent guidance could drive broader adoption efforts across pharma company silos and organizations, offering meaningful tailwinds for suppliers if these efforts translate into real change.