Introduction
A Health Technology Assessment (HTA) aims to determine the value of a health technology in a given context, with the ultimate purpose of informing decision-making on reimbursement and pricing. In Europe, HTAs are conducted mostly at the national level: HTA approaches, including value frameworks, data requirements and timing, differ by country. The country- specific setup duplicates efforts and costs and might lead to delays and/or inequalities in patient access between countries, so in recent years, cross-country initiatives have begun to emerge to streamline decision-making (e.g. the Beneluxa initiative).
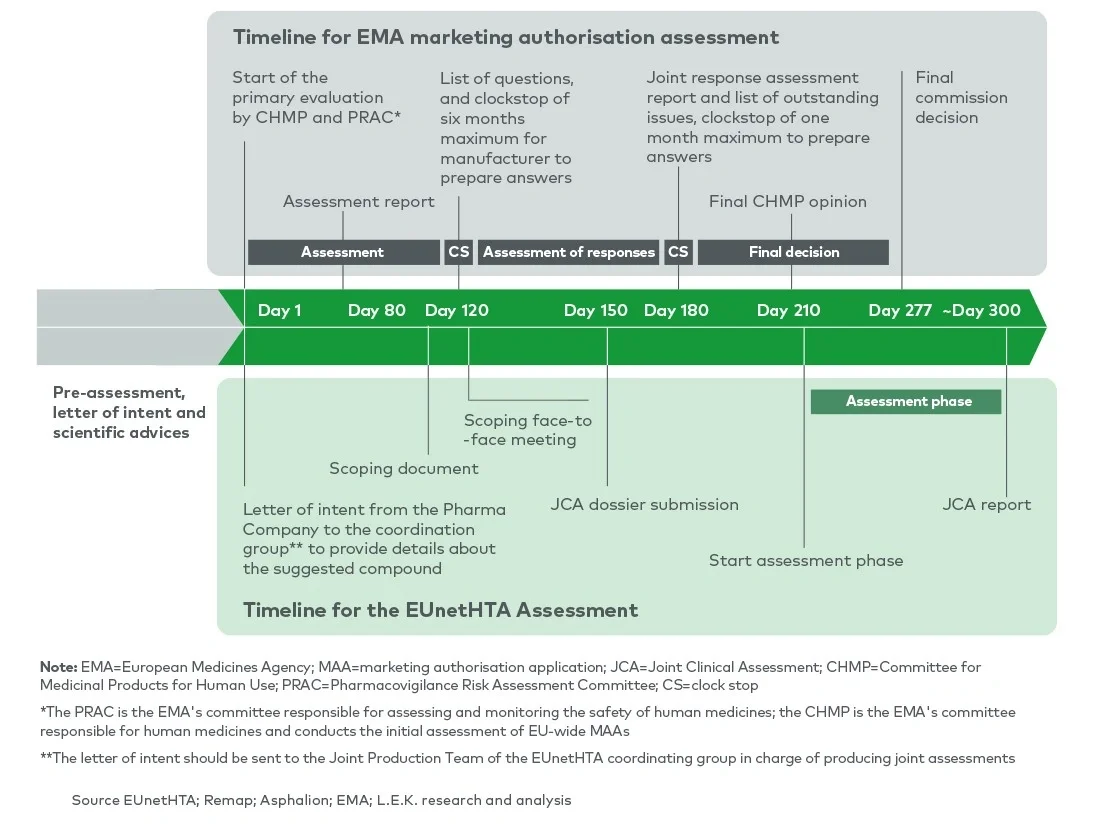
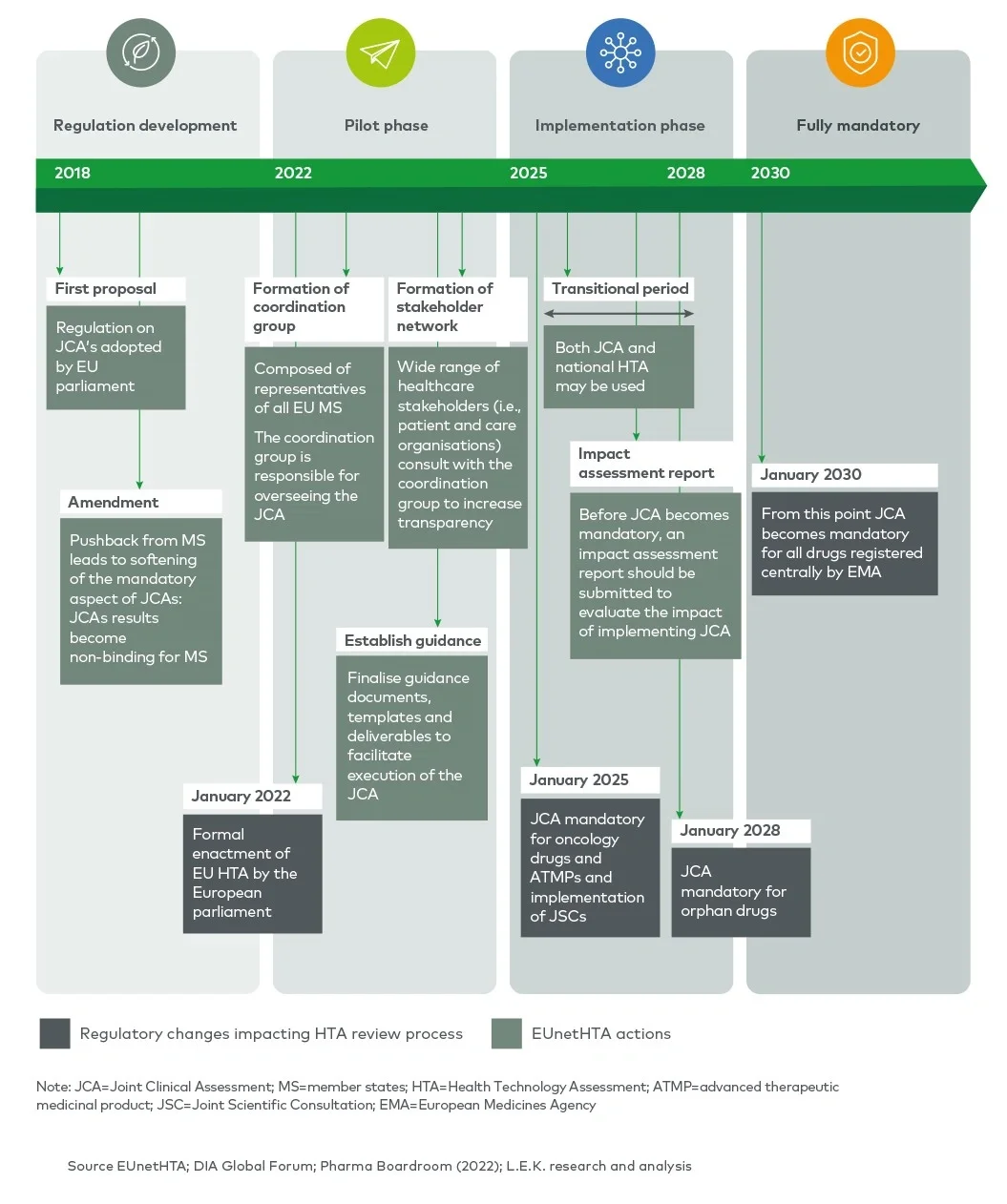
In order to improve the efficiency and quality of HTAs and increase transparency of HTA requirements, the EU HTA regulation, adopted in 2021, is seeking to create joint clinical assessments (JCAs) between member states and provide joint scientific consultations (JSCs) to applicants. JCAs will gradually become mandatory for relevant health technologies during the 2025-2030 period and will partially replace national HTAs for transferable (clinical) dimensions. JSCs will be offered to enable a dialogue between the applicant and the HTA agencies involved in the EUnetHTA network to provide early, non-binding scientific advice.
Substantial changes to the current approach to HTAs are expected from JCAs and JSCs, transitioning from the parallel interactions with national HTA bodies to a mix of EU-wide and national interactions. This Executive Insights outlines the key changes and implications for pharma companies.
Fifteen years of EUnetHTA work culminating in the EU HTA regulation 2021
EUnetHTA (European Network for Health Technology Assessment) was incepted after the European Commission and Council of Ministers recognised an “urgent need for establishing a sustainable European network on HTA” in 2004. Initially established with 35 partners, it grew to 82 national, regional and not-for-profit agencies from 29 EU member states and the UK.1 The main goal of EUnetHTA is to provide a platform for HTA agencies across Europe to exchange HTA information and develop HTA methodologies in order to ultimately facilitate the harmonisation of HTA approaches across member states. In 2009, the initial EUnetHTA collaboration was organised into an operational network, and tools to harmonise HTA assessments (e.g. the HTA Core Model®) were produced. From 2010 to 2021, EUnetHTA focused on three sequential joint actions (JA1, JA2 and JA32), resulting in the development of HTA principles, tools and methodological guidance, and in the creation of a permanent cross-border HTA working structure which connects national HTA agencies, research institutions and health ministries.
EUnetHTA actions resulted in the adoption of the EU HTA regulation in 2021, which sets out the regulatory basis for JCAs and JSCs across EU member states (EUnetHTA 21).
JCAs as a non-binding assessment of clinical domains
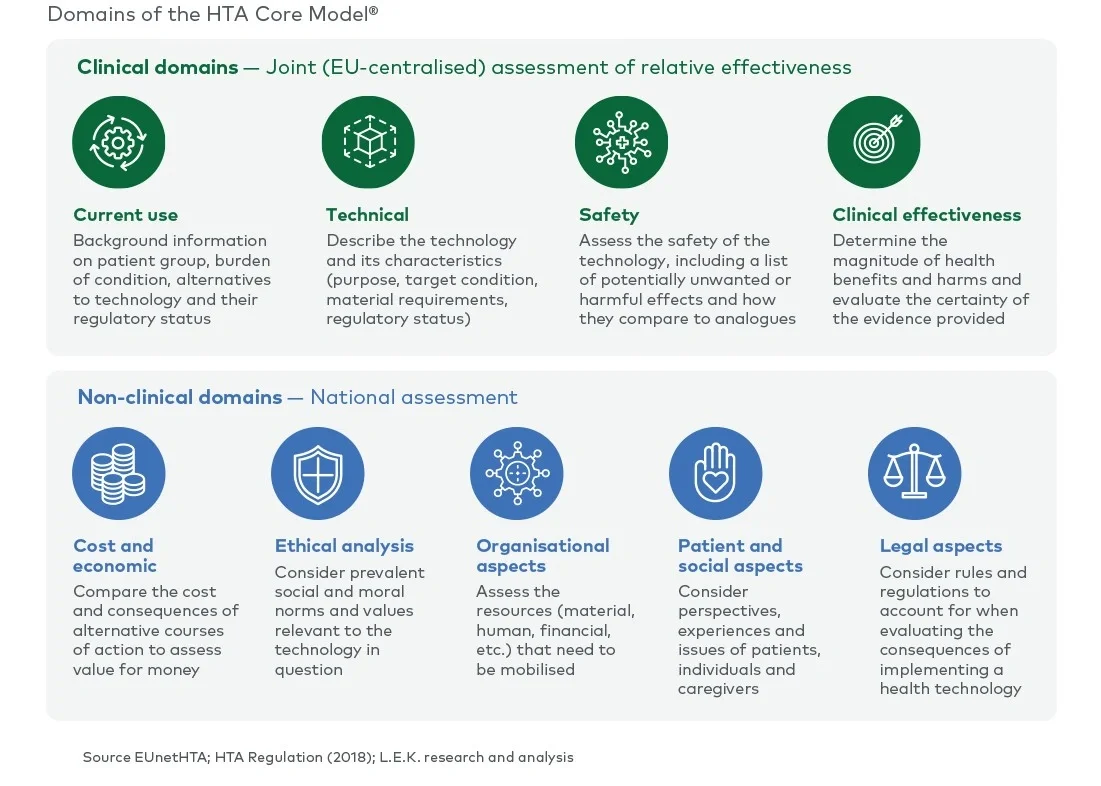
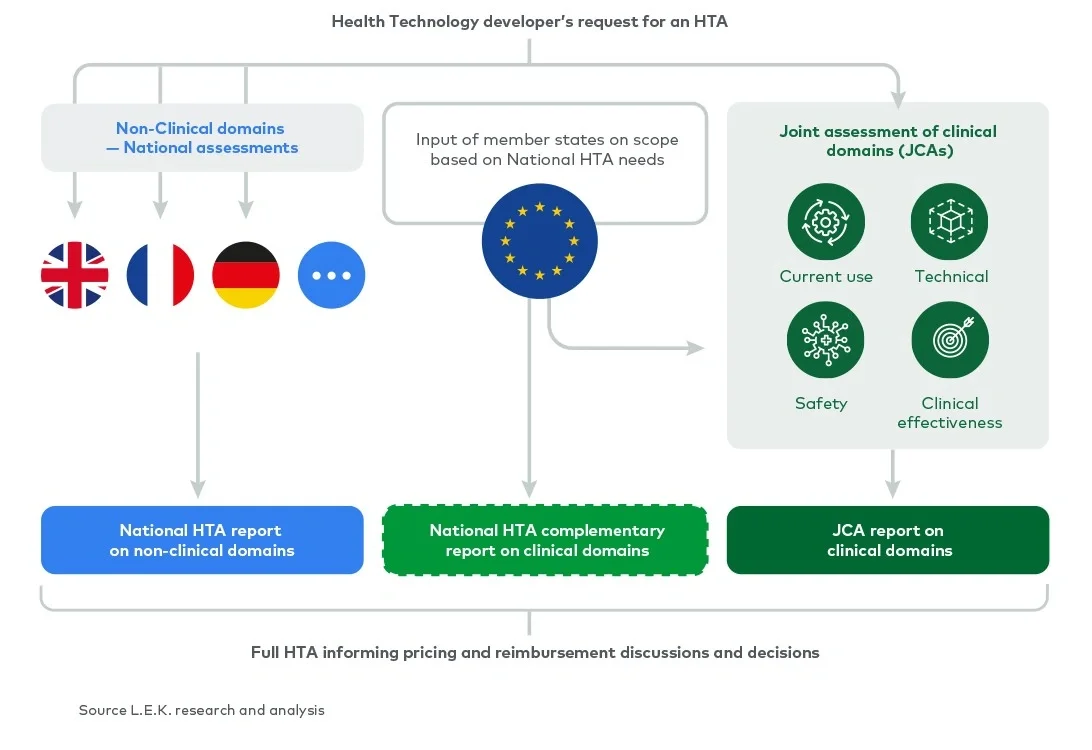
The scope of a full HTA covers the assessment of nine domains (see Figure 1). Currently, all nine domains are assessed nationally by each member state. Under the EU HTA regulation, four (clinical) domains are considered transferable across borders and will be assessed at the EU level as part of a JCA. The remaining (non-clinical) domains will still be assessed by each member state in the context of its national policies and economic resources.