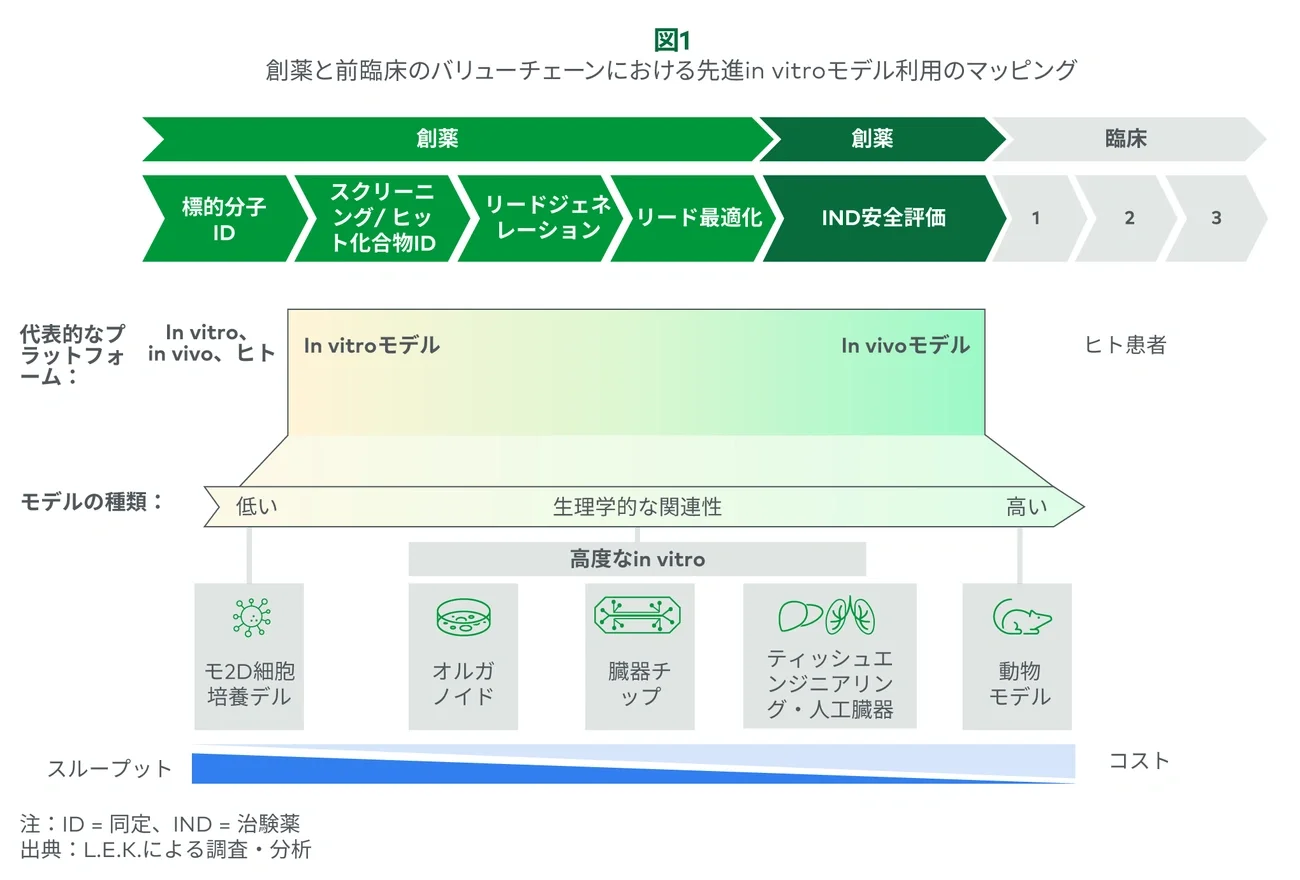
1. 探索・前臨床ワークフローにおける in vitro / in vivo の既存モデルは強固に根付いている
In vitroとin vivoモデルは、長年、医薬品開発の基盤をなしてきました。決して最適な方法ではありま せんが、信頼性と実用性に優れ、臨床的効果の高い医薬品の開発を支え続けてきたことがその理由で す。In vitroは、一般的に2D細胞培養モデルで構成されています。これらは単純なモデルであるものの、製薬会社は単一リードアウトのアッセイでハイスループットフォーマット(384ウェルプレート)を活用 し、大規模な化合物ライブラリの有効性や安全性の評価を行っています。これらの2D細胞培養モデルのアッセイでは、多くの場合、疾患(例:乳がん)の再現や、安全性試験(例:hERGのイオンチャネル)が主に行われます。先進in vitro系は、より高い精度が期待できます。しかし、スループットが高い2Dモデルが広く定着していることが採用への高い障壁となっています。
さらに、先進in vitro系は費用が高いため、精度の高さなどによる優位性の観点だけでは正当化できな い可能性があり、実用性が重要な検討材料となります。このところサプライヤーは、単純な2Dモデルでは満たすことができない複雑な疾患領域(例:がん免疫療法や筋ジストロフィー)や、高度なモデルにより以前よりも高いスループットが可能になった領域に重点的を置いています。スループットの向上と疾患発症メカニズムへの洞察を深めるために、高度なモデルへのAI統合がますます進んでいます。AIと機械学習 (ML)のアルゴリズムは、複雑なデータの簡素化や、早期における有効性と毒性のパターンの特定に役立ちます。
In vivoげっ歯類モデルは、一般的にリード化合物の最適化から始まり、生理学的に関連性のあるシステムで新薬候補を評価するために用いられます。これらのモデルは、一匹から複数のリードアウトが得られることが多く、一連の試験(安全性、有効性、ADME)に活用されます。
例えば、用量の漸増試験では、肝臓、腎臓、心臓などの臓器の毒性評価や病理学的評価を行うこともできます。
先進in vitroモデルはin vivoモデルと同等の予測性を得られる可能性があります。しかし、生理学的複雑さや生理学的状況(例:多臓器系)の欠如により、広範な利用には限界があり、研究者がin vivoモデル から置き換えるのを妨げてきました。動物モデルは、リソース集約型で橋渡し研究の価値が限られます(例:非ヒト霊長類)が、先進in vitroモデルへの入り口と見なされてきています。最近サプライヤーは、特定の単一リードアウト(例:遺伝子治療、力価の最適化)に多くの動物が使用されるユースケースに狙いを定めています。
サプライヤーは、これらのモデルにAIやMLを組み込むことにより、モデルのベクトル化能力、カプシド設計の最適化予測、導入効率の理解の分野で研究者をサポートしています。AIと高度なシステムは、遺伝 子治療以外においても、心毒性や免疫反応などの複雑で予測性が不十分な領域で動物使用の削減に役立っています。このことは、先進システムが、FDAが目標とする動物実験の削減、さらには廃止の実現に向けた重要な出発点となる可能性を示しています。
2. 先進in vitroシステムは、完全に検証されるまでは追加コストが掛かる可能性が高い
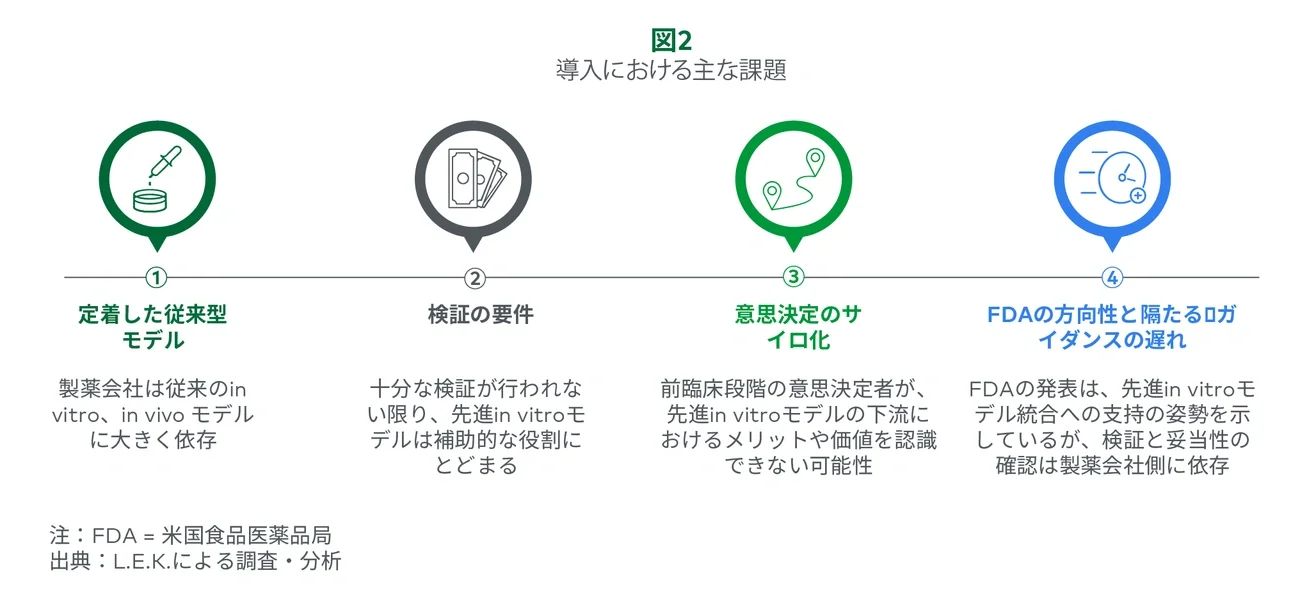
現在受け入れられている従来のモデルから先進in vitroモデルへの移行について説得するにあたり、検証が重要な鍵となります。しかし、これは未だに高いハードルとなっています。十分な検証ができない限り、研究者は従来の実験を続けなければなりません。つまり、先進in vitroモデルは代替モデルではなく追加コストとなることを意味します。製薬会社のステークホルダーは、回顧的バリデーションと予測的バリデーションを要求するケースが多くあります。回顧的バリデーションでは、先進in vitroモデルにおいて類 似の特異性と感度が予測できることを示すために、サプライヤーは既知の毒性プロファイルを持つ治療薬を活用します。臨床試験で失敗した治療薬のうち、従来のin vitro、in vivoモデルでは毒性がないとみなされていても、他のモデルでは毒性が検出されたケースがあります。
ステークホルダーはまた、他の研究者が先進in vitroツールをうまく活用し、医薬品が臨床試験へ進み、最終的には承認取得につながるための後押しをする予測的バリデーションも求めています。回顧的バリデーションと予測的バリデーションは、高額な先行投資と複数の推進者による早期の導入が必要であ り、結果としてコストが掛かります。.
現在のコストと時間の制約がある環境の中において、とりわけ既存の実験を置き換えるものではない研究を正当化することは難しい状況にあります。これらの追加コストを正当化することにおいて、サプライヤーは依然として逆風にさらされています。広範にわたる検証がない限り、先進in vitroモデルは開発ワークフローの付加的な役割に留まる可能性があります。
この状況を打開するために、サプライヤーは価値提案を磨き上げ、実用性を明確に明示して推進者を増やして行かなければなりません。規制当局への信頼性と商業利用を高めることを目的として、既知の臨床試験結果に対して、モデルの検証やベンチマークのためにAI/MLを活用するサプライヤーが増えています。AI/MLアルゴリズムは、高次元データ(例:トランスクリプトミクス、表現型スクリーニング)を分析して、In vitroモデルが関連する疾患生物学や薬剤反応と一致していることを証明することができま
す。FDAの最新のガイダンスにより、高度なin vitroモデルのアプローチの周辺で幅広い協業体制が加速され、信頼が築き上げられ、機運が高まる可能性があります。
3. 先進in vitroモデルの価値は下流で蓄積され、購買関係者まで届かない可能性がある
創薬と前臨床のチームは、臨床チームとの間でサイロ化が存在することが多く、先進in vitroモデルの価値を十分に引き出すことができません。この連携不足により、購買チームはPTRSの向上など、早期の適切な意思決定がもたらす下流におけるメリットを認識できない可能性があります。例えば、創薬研究者 が肝毒性プロファイルを持つ新薬候補を除外するのにオルガノイドモデルが役立つ場合、肝障害リスク の低いリード候補物質を前臨床試験や臨床試験に用いるメリットが気付かれないまま進む可能性があります。
チーム間での可視性の低さのために、初期段階において失敗した創薬の影響が十分に認識されない可能性があるため、コストと時間の節約にも影響が出ます。新薬候補がバリューチェーンのある段階から次の段階が移る時に、これらの効率改善やコスト削減が見過ごされることもあります。さらには、「早期に候補から外す」ことが常に奨励されているわけではありません。研究者の評価は、新薬候補の数を指標とすること多く、必ずしも下流における成功に基づくものではないためです。
先進in vitroモデルの価値を示すために、サプライヤーは、ツールが重要な意思決定(例:初期段階における安全性や有効性の示唆に基づき候補物質の優先度を下げる)にどのように役立つか追跡し、生産 性の向上(例:時間やコストが掛かるin vivoモデルと比較して)について強調していかなければなりません。それぞれの臨床資産には複数のモデルと数百にもわたる実験が用いられることがある一方で、サプライヤーは、先進in vitroの各モデルの価値提案に頼りながら製薬業界にその効果を納得させなければならず、従って、これらのプロセスは困難を伴います。
4. FDAによる最近の発表は前進を示しているが、広い範囲で動物モデルを置き換えるのには時間と慎重なプロセスが必要
従来のin vitro、in vivo試験と比べ、先進in vitroモデルを支持する証拠が増えてきているにもかかわらず、FDAはこれらの技術を全面的に受け入れ、規制枠組みの中に取り込むのに時間がかかっています。FDAはここ数年、ガイダンス文書の発行、法案策定(例:FDA近代化法2.0)、適格性評価プログラムの着手(例:ISTAND)、共同研究支援など段階的に進めてきました。ただし、採用ペースは慎重です。
FDAの最新ガイダンスでは、動物使用の削減と代替モデルへの移行に関してより積極的な姿勢が示されています。しかし、mAbの霊長類実験要件の段階的廃止以外、ロードマップは依然として要望のレベルです。限定的に既に活用されているアプリケーション(例:肝臓臓器チップ)を除けば、先進in vitroの領域は発展前の段階にあり、新たなモデルの多くは検証や出資者からの信頼が不足しています。FDAは、ボディ・オン・チップのシステムや薬物動態向けAI/MLモデルなどの初期段階の構想を強調しています が、規制側の期待や導入への具体的な道筋については明らかにしていません。
FDAはより予測可能なツールへの移行を支持する姿勢ですが、現在のところ、検証や規制の根拠に関する負担は製薬会社側に掛かっています。そのため、製薬会社側は、FDAがオルガノイドや臓器チップシス
テムからのデータを実際に受け入れるかどうかを見極めなくてはならない状況にあります。加えて、FDAと製薬業界は、見慣れない毒性データを使った研究を行うことに消極的な臨床チームなど、他の関係者にも説明しなくてはなりません。
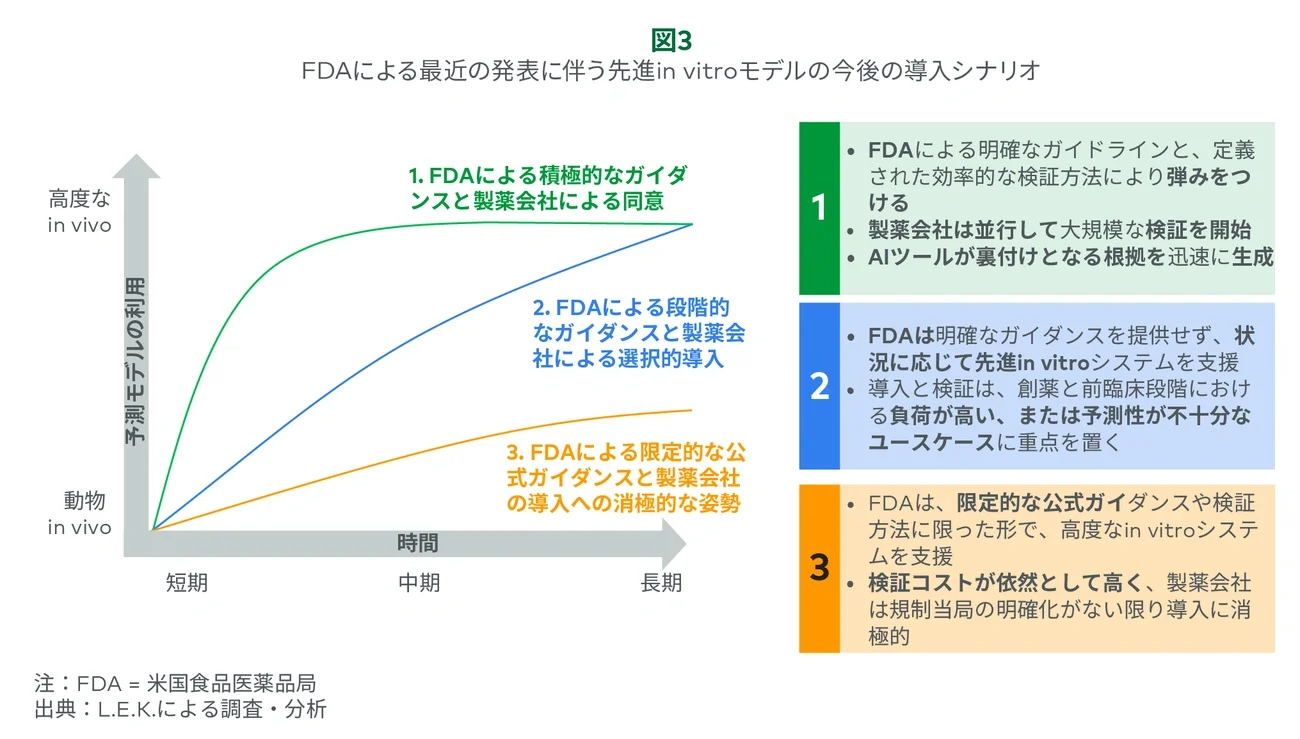
そのような状況にあっても、このガイダンスは転換点としての大きな意義を示しています。短期、中期、長期にわたる業界と規制側の対応により、導入が加速的に進むか、または遅いペースで進むのかが決まります(図3参照)。製薬会社は、規制側の認識を変えるために必要な検証データを生成することにおいて重要な役割を果たします。これは特に、従来のモデルでは高コストの上、予測性が不十分な優先度が高いユースケースにおいて重要です。効果的な導入には組織的な検証、より明確な期待、また高度なin vitro、in silico、AI/MLモデル全体へのサポートが必要です。これらを進めることができれば、FDAのガイダンスは、製薬会社の壁や組織を超えた幅広い導入の取り組みにつながります。そして、この取り組みが真の変化をもたらすことになれば、サプライヤーには意義のある追い風となります。